文章信息
用于表征植物免疫受体库的靶向长读长测序方法的比较分析
Comparative analysis of targeted long read sequencing approaches for characterization of a plant’s immune receptor repertoire
发表期刊:BMC Genomics
影响因子:3.730
发表单位:东英吉利大学环境科学学院
技术手段:MinION及PacBio RSII长读长测序(三代测序)

R基因(Plant resistance-genes,植物抗性基因)是通过育种控制植物疾病的宝贵资源:通过杂交或转基因策略引入抗性等位基因,可以使作物对病原体具有抗性。植物育种非常耗时,成功与否取决于含有所需R基因序列的植物相容性的可用性。这些方法的应用有限,并且病原体进化速率可能超过产生抗性植物品种的速率。
在植物基因组中,NLR(nucleotide-binding leucine-rich repeat,核苷酸结合位点富含亮氨酸重复序列)编码基因可以出现在具有几乎相同序列的多个基因簇中。最近的几项研究报道了用于表征和克隆植物R基因的改进方法。Witek等人使用抗性基因富集测序(RenSeq)结合PacBio RSII长读长测序克隆马铃薯晚疫病的抗性基因。基于PacBio RSII的RenSeq,即SMRT RenSeq,能够靶向捕获NLR基因和邻近的基因间和基因内区域的整个编码序列,从而提升这些簇内相似NLR基因的差异。
作物的选择性育种导致遗传多样性减少,野生近缘种通常是通过NLR免疫受体编码的基因对病原体产生新的遗传抗性的关键。最近的研究已经证明了作物NLR谱如何通过Illumina或PacBio(RenSeq)测序平台进行靶向测序以及鉴定传递病原体抗性的特定基因。随着SMRT RenSeq在植物R基因克隆中越来越受到关注和应用,关于Oxford Nanopore Technologies(ONT)公司的MinION™测序仪是否能在这个方面有所应用还未见报道。
本研究利用Nanopore测序(ONT MinION R7.3试剂)重复了Witek等人的SMRT RenSeq实验,测试了序列组装方法,并将MinION的测序结果与已报道的PacBio RSII数据结果进行了比较。最后通过计算机实验检测MinION是否能够从样品中鉴定新的NLR基因。
项目流程1. 目标序列捕获扩增,
2. MinION测序文库构建及测序,
3. 原始纳米孔单分子数据处理,
4. 预装配Read过滤,
5. 脱靶率确定,
6. Minion 2D pass Read和PacBio Read插入Canu组件,
7. HGAP组装,
8. Read和Contig质量控制,
9. NLR蛋白质Motif分析,
10. MUMer组装比较,
11. NLR蛋白质序列预测,
12. Pilon MinION Contig修复,
13. 新型NLR基因预测。
为了创建一个与已发表的SMRT RenSeq可比较的MinION数据集,本研究扩增了与先前研究中光果龙葵相同的目标DNA区域 ,并构建了两个测序文库。将测序数据结果与之前的PacBio数据进行了比较。
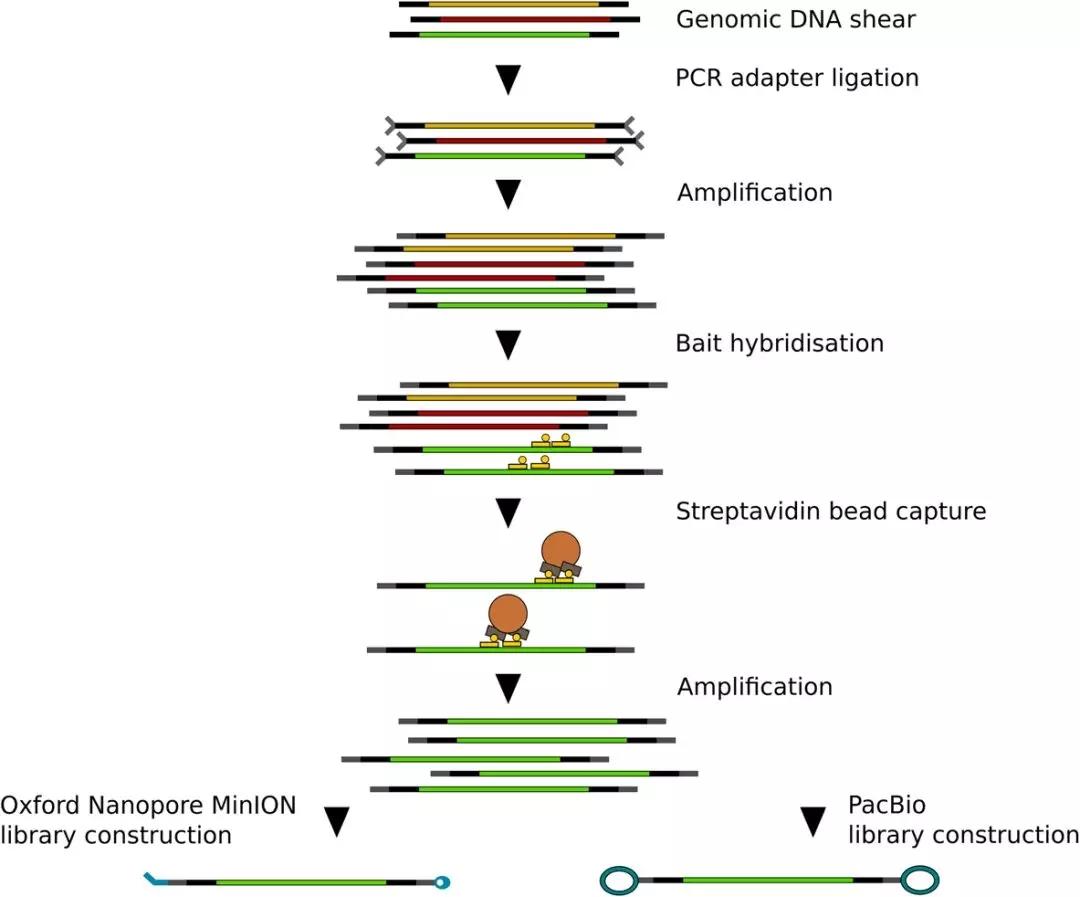
图1 MinION文库及PacBio文库
1.Read质量比较
研究评估了failed 和 passed的MinION Read以及PacBio SR和RoI的数据质量。 结果表明MinION 2D pass Read的模态精确度为92.06%,平均精确度为91.36%,与PacBio SR(模态SR准确度为90.00%,平均SR精度为89.89%)更相似,而PacBio RoI模态精确度为 99.99%,平均准确度为99.57%。研究还发现MinION数据的插入序列较短,如MinION 2D pass Read长度为2.8kb低于PacBio插入序列长度(3.5kb)。由于MinION 2D pass Read是最准确的MinION序列类型,而PacBio RoI是最准确的PacBio序列类型,因此以下分析仅基于MinION 2D pass Read和PacBio RoI。
表1 ONT MinION R7.3和PacBio RSII测序性能的比较
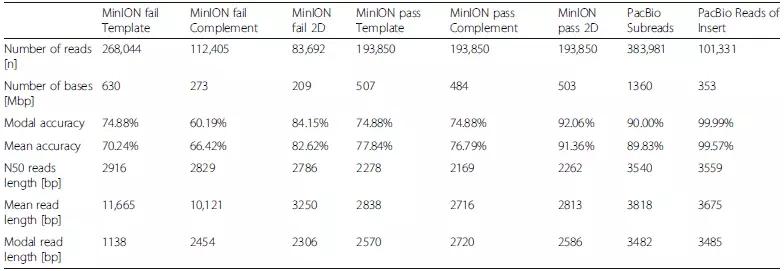
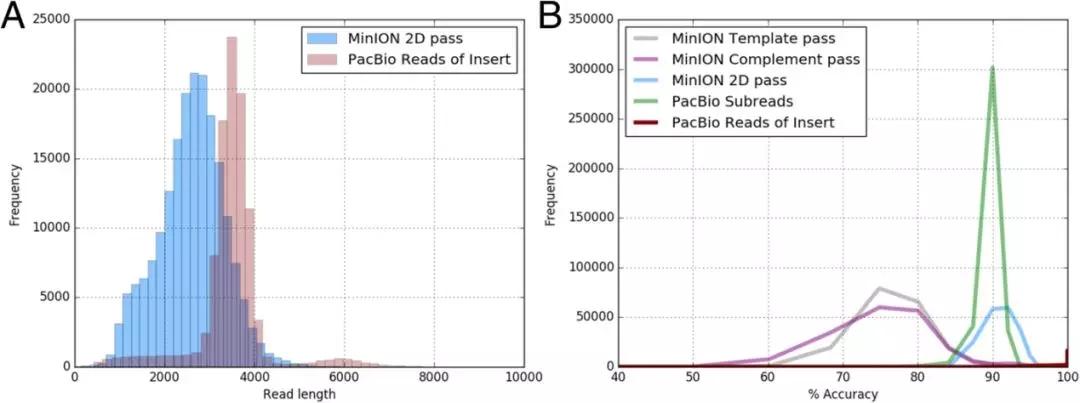
图2 ONT MinION和Pacbio RSII测序平台之间的性能比较
2.Reads处理和组装
为了确定目标序列捕获率,计算了包含96个碱基的baits序列的reads。利用BLAST评估了reads和baits之间的序列相似性。校正的MinION 2D pass和 PacBio RoI 分别包含81.28%及73.73%诱饵序列。对于PacBio SR和MinION 2D pass reads,这个比例较低(未校正的MinION 2D pass为62.50%,PacBio SR为57.82%)可能是由于较高的Read错误率的影响。
为了评估Read数据中NLR基因的百分比,使用NLR Parser软件基于它们的motif预测NLR基因。由于该软件对indel非常灵敏,仅使用经过校正的MinION 2D pass Read(49.30%序列包含NLR蛋白motif,其中10.32%为完整序列)以及PacBio RoI数据(45.41%包含NLR基序,其中13.83%列为完整序列)进行预测。PacBio RoI数据中较高数量的完整注释Reads与较长的插入序列大小一致。
表2 组装数据及NLR-Parser组装评估
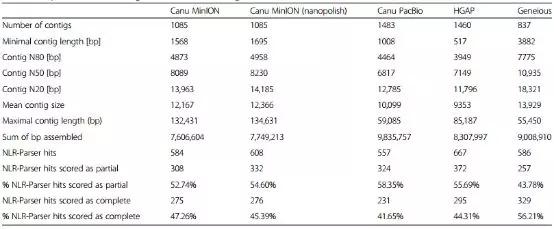
随后,利用Canu组装了经过校正的MinION 2D Read和PacBio RoI。在组装MinION 2D pass Read后,用纳米蛋白进一步校正contig。此外,研究还使用对PacBio RenSeq 数据进行修饰的Pipeline软件对PacBio RenSeq数据进行了HGAP组装。结果获得了纳米抛光的MinION 2D pass Canu组件组装的1085个contig(7.74 Mb),PacBio RoI Canu组件组装的1483个contig(9.14 Mb),PacBio HGAP组件组装的1460个contig(8.31 Mb)和用于PacBio Geneious组装的837个contig(9.01 Mb)。Canu和HGAP组装的N50值均短于Geneious组装的N50长度。Canu组装的平均contig大小与Geneious组装的相当。
研究使用NUCmer将所有组装与Geneious参考序列对齐(最小长度设为500),并使用mummerplot显示对齐。在所有组装中,纳米抛光的Canu MinION 2D pass组装最接近于手动校正的Geneious参考序列。通过对数据进行纳米抛光,Canu MinION组装与参考序列之间的一致性显着增加(图3)。
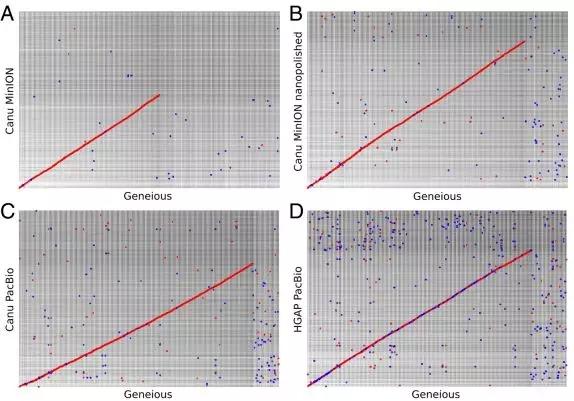
图3 NUCmer 组装比对与Geneious的比较
3. 组装的NLR编码基因的评估
为了计算NLR编码Contig的数量,本研究利用NLR-Parser分析了装配情况,还使用AUGUSTUS分析了编码NLR蛋白的装配,从而预测蛋白质序列,然后使用NLR-Parser来鉴定NLR蛋白。结果表明,使用高度准确的PacBio RoI数据生成的装配体在蛋白质预测分析中比Canu MinION装配表现更好。此外,研究还发现HGAP PacBio装配预测到的蛋白质与Canu PacBio装配相比相似度更高。
组装的Canu MinION contigs中的错误可能源于富含indel的MinION测序误差分布。由indel引起的移码突变可导致错误或无从头蛋白质的预测,这可能是导致PacBio和MinION数据之间差异的原因。通过用高质量Read进行contig校正可以实现组装效果的改进,如插入indel低的短Illumina测序序列。研究结果表明增加MinION数据质量,例如用Illumina序列数据进行contig修饰有利于预测蛋白质编码基因。
表3 AUGUSTUS 蛋白质预测结果
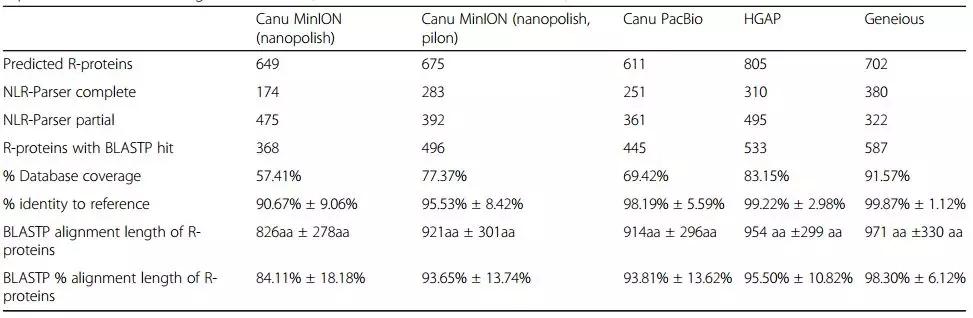
使用快速有效的工具如BLASTN和NLR-Parser鉴定新的NLR基因是非常有效的。本研究利用BLAST对获得的contig进行搜索,得到了649个NLR基因参考数据库。74个MinION和19个PacBio contig映射到完整的NLR基因参考数据库。通过手动检查contig与NLR基因序列的比对,对于MinION数据,恢复了37个融合域NLR基因中的29个,平均同源性为98.00%±0.91%,平均比对长度为6060bp±1661bp。对于PacBio数据,恢复了37个融合结构域NLR基因中的16个,平均同源性为99.57%±0.47%,比对长度为5569bp±1272bp。
MinION和PacBio数据之间组装的融合NLR基因的差异可能源于进入装配通道中较大数量的MinION Read数。换句话说,PacBio RoI Read,由于其较高的质量,与相关序列比对分值较高,因此不考虑组装,而更多的MinION Read进入组装(质量较低和BLAST得分较低的read)则不会被过滤掉。表明对于这种类型的分析,尽管NLR基因的序列相似性很高,但MinION数据表现良好。
表4 adapter curation前后Read统计信息的比较

通过对ONT MinION测序平台的测试,其在性能上可以替代PacBio测序平台。这使得MinION成为重复富集基因组(基因组区域包含相似基因簇,如NLR基因)的有效工具。本研究结果表明MinION在长读长测序上可以与PacBio RSII竞争,并发现在R7.3 flow cell上MinION 2D pass的准确性与PacBio SR相当。
为了将NLR基因组装到99%以上的准确度,本研究提出了MinION 2D pass的装配策略,其基于cutadapt处理,使用BLASR的嵌合读取过滤,Canu读取校正和修剪,并使用纳米抛光进一步抛光组装的contigs。研究结果表明,与手动注释的PacBio生成的参考数据相比,MinION contigs的精确度为99.41%。此外,研究结果还表明,Canu MinION组装的NLR基因预测的蛋白与SP2271 NLR基因参考的预测蛋白质之间的同源性较低(90.67%)。
研究结论研究结果表明与PacBio SMRT cell 相比,ONT R7.3 flow cell平台测序产量较低,但MinION的读长和Canu MinION 2D pass组装的精确度表明了ONT MinION用于研究感兴趣的基因组区域的有效性。在不久的将来经过改进的ONT测序技术将允许检测更加复杂的基因组区域,包括原核及真核生物,如植物和人类。“Read Until”等软件应用程序的开发可以进一步将ONT平台推进到基于可调生物信息学的富集性靶向测序的独特测序设备。
参考文献Matthew D. Clark,Michael Giolai,et al. Comparative analysis of targeted long read sequencing approaches for characterization of a plant’s immune receptor repertoire.BMC Genomics,2018.
拓展阅读